개요
앞서 단백질의 특이성(specificity)에 대해 이야기하였다. 세포 내에서 일어나는 이러한 단백질의 특이성은 마치 단백질에 눈이 달린 것 처럼 작동한다고 생각하기 쉽지만, 단백질의 특이성 또한 결국 물리화학적 법칙을 따르는 분자의 운동에 불과하다.
즉, 단백질 분자가 외부 계의 물리적 힘 - 특히 열 - 에 의해 진동하거나 부유하는 brownian motion을 통해 돌아다니다가, 적절한 DNA 서열과 유효충돌이 일어나야 상호작용이 일어나는 것이다.
따라서, 우리는 다음과 같은 가설을 세울 수 있다.
1. DnaA box가 많을수록 DnaA protein과의 유효충돌 횟수가 커진다.
2. DnaA box가 많이 존재한다면, 이 서열들 중 일부에 변이가 일어나도 결합을 방해하는 데에 미치는 영향을 줄일 수 있다.
그러므로, 위 가설에 따르면 문제는 반복되는 특정 서열을 찾는 것으로 귀결된다. 하지만 여전히 우리는 이 특정 서열 의 길이가 어느정도인지 알지 못하기 때문에, 임의의 길이를 가지는 특정 서열을 k-mer로 정의한다.
k-mer: 길이가 k인 문자열
이렇게 k-mer pattern을 임의로 정한 뒤, 주어진 입력 Text에서 얼마나 등장하는지 그 횟수를 count 할 수 있다.
가장 간단한 방법으로, 다음과 같이 모든 각 문자열을 시작점으로 하는 k-mer와 비교할 수 있다.
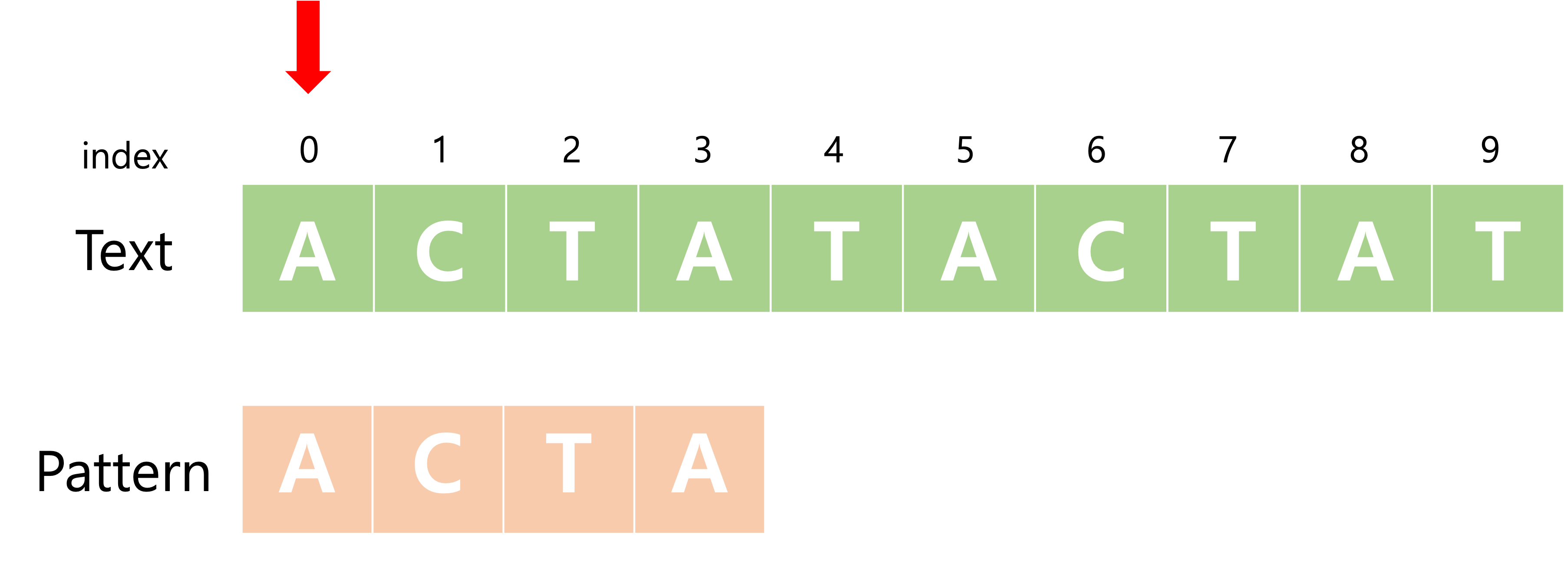
Fig 1. index가 0일 때 주어진 text에서 pattern(ACTA)찾기

Fig 2. index가 1일 때 주어진 text에서 pattern(ACTA)찾기
Problem
- Input: 전체 문자열 Text, Text에서 찾으려는 문자열 Pattern
- Output: Text에서 등장하는 Pattern의 횟수
- function: Pattern $\to$ count
Pseudo-code
PatternCount(Text, Pattern)
count <- 0
for i <- 0 to |Text| - |Pattern|
Pattern' = Text(i, |Pattern|)
if Pattern' == Pattern
count <-- count + 1
return count
Evaluation
Time Complexity
이 알고리즘의 경우 Brute-force1 방식으로, 모든 경우의 수를 순차적2으로 확인한다.
입력 크기: $\left\vert Text \right\vert = n, \left\vert Pattern \right\vert = k$
$\text{Constraints: n ≥ k}$
- line[1]: Pattern이 등장하는 횟수를 저장할 변수 선언3 $\to O(1)$
- line[2]: Text와 Pattern의 입력 크기를 각각 $\left\vert Text \right\vert$ , $\left\vert Pattern \right\vert$ 이라 할 때, Text 문자열에서 Pattern 길이의 부분 문자열(substring)을 형성할 수 있는 모든 시작점은 0번째부터 $\left\vert Text \right\vert - \left\vert Pattern \right\vert $ 번째까지 이다. → $O(n-k)$
- line[3]: 현재 순번에서 가능한 부분 문자열 형성 - $Text(i, \left\vert Pattern \right\vert)$ 만큼을 slicing 해오는 연산 $\to O(1)$
- line[4]: sliced substring과 Pattern의 비교 연산 → $O(k)$ 4
- line[5]: count 변수의 산술 연산 → $O(1)$
- line[3] ~ line[5]의 경우 상수항의 시간이 소요된다.
- Worst case: $n » k \to n-k \approx n$
Total Time Complexity: $O(1) + O(n-k) \times (O(1)+O(k)+O(1)) \approxeq O(nk)$
Implementation
# 전체 Text에서 주어진 Pattern의 등장 횟수를 반환하는 함수
def PatternCount(text, pattern):
# 결과로 반환할 변수 선언
count = 0
# pattern의 시작점이 될 수 있는 영역을 모두 순회
for idx in range(len(text) - len(pattern) + 1):
# 현재 인덱스를 시작으로 하는 pattern이 입력 pattern과 같다면 count
if(text[idx:idx + len(pattern)] == pattern):
count += 1
return count
Discussion Points
Summary
- 주어진 유전체에서 어떤 특징을 찾음으로써 특이성에 대한 단서를 얻을지도 모름
- 이러한 특징에 대한 접근으로, 유전체에서 빈번하게 나타나는 패턴을 찾는 접근을 시도해 볼 수 있음
Implementation Strategy
이러한 빈번한 패턴을 찾는 문제를 해결하기 위해 다음과 같은 전략을 통해 코드를 구성하였다.
- 주어진 전체 text에서 가능한 모든 substring의 조합을 확인함
- 가능한 모든 조합의 substring을 주어진 pattern과 일치하는지 확인하여 일치할 때마다 그 count를 셈.
Implications
- 유전체 상에서 임의의 서열이 얼마나 등장 하는지 확인할 수 있게 되었다.
- 이러한 임의의 서열은 DnaA box와 같이 ori 를 구성하는 주요 서열의 후보군 으로 생각할 수 있다.
- 하지만, 이 방법만 가지고는 유전체 상에서 “어느 위치에 반복적인 서열이 있는지” 를 확인하기는 어렵다.
다음 문제에서는, 이번에 구현한 함수를 통해 가장 많이 등장하는 단어가 어떤 단어인지 확인하는 방법에 대해 알아보도록 한다.
Reference
- Compeu, P., Pevzner, P. (2018). Bioinformatics Algorithms 3/e. 에이콘 출판사
- Craig, N., Cohen-Fix, O., Green, R., Greider, C., Storz, G., & Wolberger, C. (2010). Molecular biology: Principles of genome function. Oxford University Press.